Clinical Development of Cyclin-Dependent Kinase Inhibitors in Lung Cancer
This manuscript focuses on the latest clinical developments of newer generation selective CDK inhibitors in lung cancer.
Ajay Dhakal, MBBS
Abstract
A dysregulated cell cycle can lead to cancer. The retinoblastoma (RB1) gene is a tumor suppressor gene and its corresponding protein Rb has an integral role in cell-cycle regulation. Dephosphorylated Rb halts the cycle while phosphorylated Rb restarts it. Cyclin-dependent kinase (CDK) 4 and 6 are responsible for phosphorylating Rb, D-type cyclins, which are expressed in response to stimulatory mitogens, are CDK activators while p16INK4a encoded by the CDKN2A gene is an innate CDK inhibitor. CDKs have a vital role in unleashing cell cycle arrest and are targeted by a class of antitumor drugs called CDK inhibitors.
Inactivation of Rb pathway is seen in more than 70% of nonsmall cell lung cancer (NSCLC). The most common event for inactivation is the loss of CDK2NA gene. Moreover, this inactivation occurs frequently in patients with mutant KRAS lung adenocarcinomas. Early-phase studies have shown that CDK inhibition can induce immediate senescence of NSCLC with KRAS mutation. In addition, CDK4 and cyclin D1 amplifications have also been associated with lung cancer, specifically squamous cell lung cancer. Thus, there has been a significant interest in investigating the role of CDK inhibitors in lung cancer. Various broad spectrum nonselective first generation CDK inhibitors encountered obstacles in clinical development due to poor pharmacokinetic profiles and toxicities. This review article will focus on the therapeutic relevance of newer generation selective CDK inhibitors which have recently progressed the furthest in clinical development in lung cancer.
Jiang Yio, MD, MBA
Introduction
Cell-cycle dysregulation is one of the hallmarks of cancer.1Various genes and their corresponding proteins, interact with each other, external stresses, hormones, and cytokines to regulate the cell cycle. Alteration in the regulatory pathways may lead to the dysregulation of cell cycle and tumorigenesis. Cyclin-dependent kinases (CDKs) are serine/threo- nine kinases that by definition require activation by cyclins or cyclin-related proteins and have been implicated in unleashing the cell-cycle arrest, and thus, they have been targeted by a class of antitumor drugs called CDK inhibitors. Excluding the related CDK1 (also known as CDC2)-like kinases, there are currently 20 members of the CDK family, several of which function in noncell cycle processes such as transcription. Various broad-spectrum, nonselective first-generation CDK inhibitors, such as avopiridol, encountered obstacles in clinical development due to a poor pharmacokinetic profile and/or toxicities. We will focus our discussion on the therapeutic relevance of newer generation selective CDK inhibitors that have recently progressed the furthest in clinical development in lung cancer.
Grace K. Dy, MD
Cell-Cycle Dysregulation in Cancer
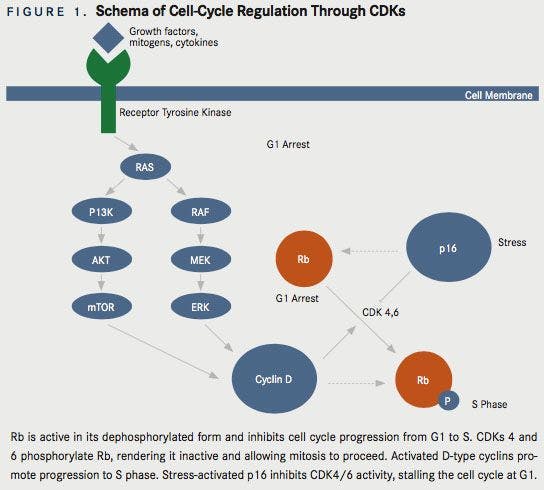
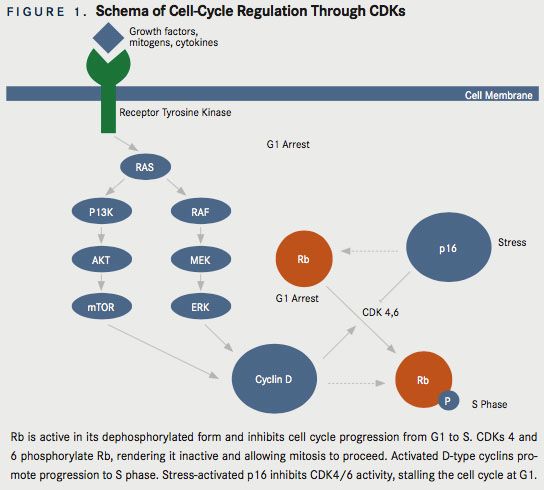
The retinoblastoma (RB1) gene is a tumor-suppressor gene that encodes for the retinoblastoma protein, pRb. The Rb protein undergoes intermittent phosphorylation as the cell traverses the cell cycle. A dephosphorylated Rb protein acts as a brake in the cell-cycle progression. Rb is dephosphorylated (activated) as the cell exits mitosis, is hyperphosphorylated (inactivated) toward the end of G1 phase, and remains hyperphosphorylated until the cell goes to mitosis phase.2CDKs regulate both the cell cycle and transcription. CDKs 1, 2, 4, and 6 are required for the correct timing and order of the events of the cell-division cycle. CDK7 is a component of the CDK-activating complex that contributes to the assembly of CDK1/cyclin B. In addition, CDKs 7, 8, and 9 function as transcriptional CDKs.3 CDK4 and CDK6 phosphorylate the Rb protein, thus releasing the cell-cycle arrest during the G1 phase. CDK4 and CDK6 are activated by D-type cyclins, which are expressed in response to various extracellular signals such as stimulatory mitogens, inhibitory cytokines, cellcell contact, and other spatial cues.2These CDKs are inhibited by phosphorylation, ubiquitination, and binding of the endogenous cellular inhibitor p16INK4a (Figure 1). p16INK4a is encoded by CDKN2A (also known as ARF or INK4a) and its expression is induced by variety of hyperproliferative stress signals.4Thus, competition between stress-activated p16INK4a and mitogen-activated D-type cyclins to bind with CDKs determines whether the cell stays in G1 arrest or proceeds to S phase.2Regulation of CDKs by p16INK4a is often disrupted in cancer cells, causing increased activity of CDKs. Additionally, p16INK4a expression suppresses tumorigenic effects in cancer cell lines.5Many studies identify the CDKN2A gene as a frequent target of inactivating mutations and deletions in many human cancers, and show that the loss-of-function alterations in genes encoding p16INK4a and pRb are mutually exclusive events in tumor cells.6There are enough preclinical data suggesting the role of CDKs in tumorigenesis. Hence, CDKs have been investigated as antitumor drug targets. CDK expression and assembly with D-type cyclins depend on the activation of the RAS-dependent kinase cascade involving the sequential activation of RAF1, MEK1, MEK2, and ERKs. Similarly, a separate RAS signaling pathway involving PI3k and AKT also prevents phosphorylation of D-type cyclins. Cancer-specific mutations affecting receptor tyrosine kinases (RTKs), RAS, RAF, PI3K, or PTEN can enhance D-type cyclin-dependent CDK4/6 activities, thus promoting oncogenesis. Conversely, inhibitors of these intracellular signaling cascades such as antiproliferative cytokines and antagonists of hormones and interleukins can decrease the activity of CDK4 and CDK6, inducing cell-cycle arrest.8
CDK4/6 inhibitors have a cytostatic effect by inhibiting the cell cycle. Thus, they are primarily expected to provide disease control rather than objective responses even though preclinical studies have shown that CDK4/6 inhibition can produce rapid tumor regression and decrease in tumor burden in multiple human tumor xenograft models at high doses. Tumor cell kill was evident with high doses of palbociclib (Ibrance), a selective CDK4/6 inhibitor against human xenograft models of colon and breast cancer in a study by Fry et al.9 However, recent clinical trials have failed to replicate objective tumor responses seen in these preclinical experiments. But, significant disease control has been shown consistently as benefits in progression-free survival (PFS) owing to the cytostatic nature of CDK4/6 inhibitors. Palbociclib demonstrated remarkable PFS benefit in the PALOMA 1 trial in combination with letrozole compared with letrozole only (20.2 vs 10.2 months) among postmenopausal women with estrogen receptor (ER)positive, HER2-negative, advanced breast cancer. There was a modest benefit in objective response (43% vs 27%).10In the PALOMA 3 trial, palbociclib in combination with fulvestrant compared with fulvestrant alone in postmenopausal women with ER positive, HER2-negative, advanced breast cancer who have progressed on prior endocrine therapy showed significant PFS benefit (9.2 vs 3.8 months). There was no significant benefit in objective response rate.11Among patients with CDK4-amplified liposarcomas that have progressed despite systemic therapy, treatment with palbociclib was associated with a favorable PFS in a phase II study. Of 29 patients evaluated, PFS was 66% (90% CI, 51-100) at 12 weeks, significantly exceeding the primary endpoint of PFS >40% at 12 weeks.12Majority of patients had stable disease (SD), with only 1 objective response noted among 57 evaluable patients. Similarly, in a phase III randomized trial, ribociclib, another specific CDK4/6 inhibitor, in combination of letrozole compared with letrozole plus placebo, yielded significant benefits in PFS. The median duration of PFS was not reached in the ribociclib group (95% CI, 19.3 to not reached) versus 14.7 months (95% CI, 13.0-6.5) in the placebo group (hazard ratio, 0.56; 95% CI, 0.43-0.72; P <.001) for superiority). There was a modest difference in response rate (52.7% vs 37.1%, P <.001).13

CDK Inhibition in NonSmall Cell Lung Cancer
Inactivation of the Rb pathway is seen in more than 70% of nonsmall cell lung cancer (NSCLC). The most common event for inactivation is the loss of the CD- KN2A gene. After multiple preclinical studies demonstrating frequent aberrations in p16INK4a among lung cancer cell lines, Gazzeri et al studied genetic changes of p16INK4a in 43 NSCLC samples.14Twenty-one of these samples exhibited one of the following genetic or epigenetic alterations affecting p16INK4a: homozygous deletion, frame-shift or missense mutation or methylation of exon 1 alpha. Moreover, its inactivation occurs frequently in patients with mutant KRAS lung adenocarcinomas. Mutant KRAS exerts its oncogenic activity through the dysregulation of several signaling networks. Among these, the most extensively characterized are the RAF/MAP-ERK kinase (MEK)/ex- tracellular signal-related kinase (ERK) and the phosphoinositide 3-kinase (PI3K)/AKT/mTOR signaling pathways.15In approximately 30% of 323 lung adenocarcinoma cases from The Cancer Genome Atlas (TCGA) database with oncogenic KRAS mutations, 53% had concurrent inactivation of p16 by one of the mechanisms described above.16Of interest, p16 methylation was linked to KRAS mutation but mutually exclusive with EGFR mutation.17Loss of p16 appears to confer a worse clinical phenotype with increased metastasis and decreased survival.16, 18Moreover, Puyol et al demonstrated that CDK4 ablation induces immediate senescence in NSCLC with KRAS-activating mutations.19Subsequent human xenograft models of NSCLC showed that tumors with KRAS mutations were more sensitive to CDK inhibitors relative to KRAS wild-type tumors.20The antitumor ef cacy of CDK inhibition against KRAS-mutant lung cancer also appears to be synergistic in combination with MEK inhibition, with combination therapy showing increased cytotoxicity than either agent alone, both in vitro and in vivo.21
In addition to activating KRAS and loss of INK4a gene causing tumorigenesis, CDK4 ampli cation and cyclin D1 (CCND1) amplifications have also been associated with lung cancer, specifically squamous cell lung cancer (SqCLC). In TCGA, CCND1 amplification was seen in 12% of SqCLC, while only 1% of SqCLC is expected to have CDK4 amplification.22Thus, there has been a significant interest in exploring the role of CDK4/6 inhibitors, specifically in lung cancer.
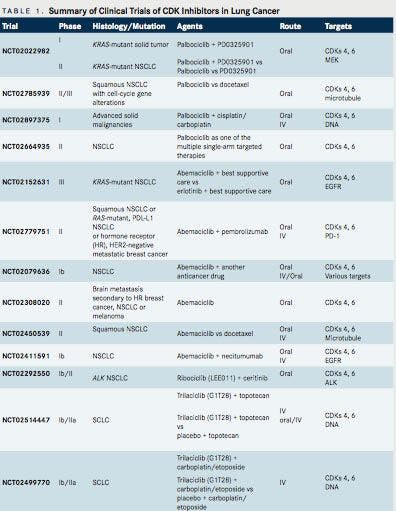
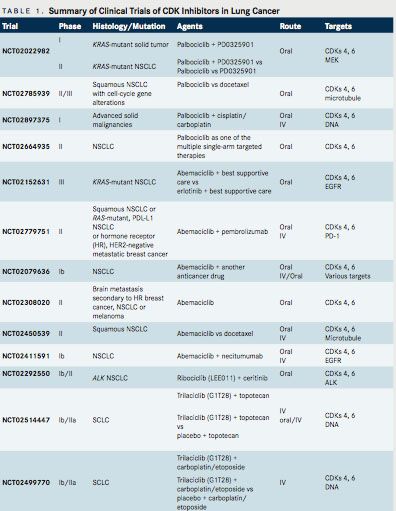
The role of CDK inhibitors in lung cancer is not as established as it is in hormone receptorpositive breast cancer. Table 1 summarizes representative trials of CDK inhibitors currently being tested in lung cancer. Palbociclib and abemaciclib (LY2835219) are selective CDK4/6 inhibitors currently being investigated in NSCLC. Palbociclib has a half-life of 25.9 to 26.7 hours and is dosed at 125 mg daily (3 weeks, 1-week drug holiday) or 200 mg daily (2 weeks, 1-week drug holiday).2Abemaciclib has a half-life of 17 to 38 hours and is dosed at 200 mg twice daily, continuously. Abemaciclib crosses the blood brain barrier. The major dose-limiting toxicities with palbociclib are neutropenia and thrombocytopenia, while those of abemaciclib are fatigue and gastrointestinal (GI) events (diarrhea).2Ribociclib is another selective CDK4/6 inhibitor that recently received priority review from FDA as first- line therapy combination with letrozole in metastatic HRpositive breast cancer. It is dosed at 600 mg daily (3 weeks on, 1 week off), is being explored in other advanced solid tumors in phase I/II trials, and has limited data in NSCLC.13
Patnaik et al evaluated a cohort of 68 patients with previously treated NSCLC, including 29 patients harboring KRAS mutations. The overall disease control rate (CR+PR+SD) with abemaciclib was 49% in the total study population, 55% in the KRAS-mutant cohort, and only 39% in the KRASwild type cohort. The 6-month PFS was 26% (95% CI, 16-38). The common KRAS mutation involving codons 12 and 13 and the less frequent KRAS mutations Q61H and A146V were associated with decrease in tumor size following single-agent abemaciclib treatment. SD lasting more than 24 weeks was seen in 31% of the KRAS mutant cohort compared with 12% in the wild-type cohort. The most common treatment-related adverse events included fatigue and gastrointestinal effects such as diarrhea, nausea, vomiting, anorexia, and weight loss. Only 1% of all the enrolled patients had grade 4 toxicity (neutropenia).20This study suggested that abemaciclib has promising activity against RAS-mutant NSCLC and that the recommended dose of 200 mg twice daily appears to be safe and tolerable.20JUNIPER (NCT02152631) is a randomized, phase III study of abemaciclib with best supportive care versus erlotinib with best supportive care in patients with stage IV NSCLC with a detectable KRAS mutation, whose disease has progressed after platinum-based chemotherapy. The primary outcome measures are PFS and overall survival (OS). The estimated study completion date is August 2019.
Gopalan et al conducted a phase II, single-arm study of palbociclib in 19 previously treated patients with recurrent or metastatic NSCLC who are negative for p16 expression by immunohistochemistry.23The primary endpoint was overall response rate. Of the 16 evaluable patients, there were no responses, thus the trial was closed to accrual. However, 8 patients with previously progressive disease had SD lasting 16 to 42 weeks. The remaining 8 patients reported progressive disease within 8 weeks. The median PFS was 12.5 weeks, comparable to other second-line chemotherapy in this setting. In addition to the modest clinical activity, 5 out of 16 patients developed grade 3 or 4 toxicities (transaminitis, rhab- domyolysis, neutropenia, and thrombocytopenia). This study required that tumors be negative for p16 expression. In contrast, the S1400C substudy of the LUNG MAP trial is investigating the role of palbociclib for patients with previously treated stage IV Sq- CLC (NCT02154490), whose tumors express gene amplification of CDK4, CCND1, 2, or 3, for eligibility. This study will evaluate the role of these cell-cycle gene alterations as a predictive biomarker for palbociclib in this patient population.
In terms of agents that inhibit CDKs other than CDK4/6, dinaciclib (MK-7965, formerly SCH 727965), a potent inhibitor of CDKs 1, 2, 5, and 9, had been evaluated in a randomized phase II study versus erlotinib in patients with previously treated NSCLC.24Preclinical studies have shown that dinaciclib is a potent inducer of apoptosis of various cell lines, including lung cancer.25, 26In this trial, 17 patients were allocated into the dinaciclib arm while the erlotinib arm had 33 patients. Dinaciclib was administered at 50 mg/m2 over 1 to 2 hours IV infusion. The study showed that dinaciclib, as expected, is more toxic in terms of myelosuppression than erlotinib. A total of 78% patients in the dinaciclib arm had grade 3 or 4 AEs, including relatively higher rates of GI events such as diarrhea, nausea and vomiting, while only 29% of patients in erlotinib arm en- countered grade 3 or 4 adverse events, mainly dermatologic. The time to progression was 1.49 months (95% CI, 1.31-2.63) with dinaciclib while it was 1.58 months (95% CI, 1.38-2.83) with erlotinib. This study did not meet its primary endpoint in establishing activity of dinaciclib as monotherapy in this population. However, unlike more contemporary CDK4/6 inhibitor studies, this study was performed among molecularly unselected, advanced NSCLC patients.
Indeed, preclinical investigations of dinaciclib suggest that its activity is exerted through inhibition of MCL1, a BCL2 anti-apoptotic family member.27Additional data suggest that high ratio of MCL1 mRNA relative to BCL- XL or high MCL1 copy number may be a predictive biomarker for antitumor response to dinaciclib in solid malignancies, including lung cancer.28Several trials are currently exploring various combinations of CDK inhibitors with other agents. As described above, MEK is the downstream protein to RAS in the MAPK/ERK pathway (RAS-RAF-MEK- ERK pathway). Activity of D-type cyclins is highly dependent on RAS signaling. Preclinical studies showing synergistic effect of CDK inhibition and MAP kinase inhibition in melanoma, pancreatic cancer, neuroblastoma, as well as lung cancer have generated significant interest in this approach.21, 29-31
Perhaps the predominantly cytostatic effects of CDK4/6 inhibition could be reprogrammed to induce apoptosis in response to drugs targeting RTK and/or downstream MAPK signaling pathways that are essential for cell viability.2Indeed, a recent screen of drug combination in ALK-mutated neuroblastoma showed synergistic efficacy for the combination of ceritinib, an oral ALK inhibitor, with ribociclib, compared with each drug alone.31A phase Ib/II study is evaluating safety and efficacy of this combination in patients with ALK-positive NSCLC (NCT02292550).
As described previously, combination of MEK and CDK inhibition was synergistic in KRAS-mutant NSCLC. A phase I study is ongoing to determine the maximally tolerated dose and recommend phase II dose for the combination of palbociclib and MEK inhibitor PD-0325901. Subsequently, the randomized phase II portion of the study will then further analyze the response rate and PFS with the combination regimen (palbociclib with PD-0325901) compared with single-agent palbociclib and single-agent PD- 0325901 (NCT02022982) for this lung cancer genotype. The estimated study completion date is December 2020.

CDK Inhibition in Small Cell Lung Cancer
Data for CDK inhibition in small cell lung cancer (SCLC) are sparse. CDK4/6 inhibitors require a functioning Rb gene to exercise their antitumor property. There is a loss-of-function mutation of the Rb gene in about 90% of SCLC, so CDK4/6 inhibitors are theoretically ineffective.32Roniciclib (BAY 1000394) inhibits the activity of CDKs 1, 2, 3, and 4, and of transcriptional CDKs 7 and 9. It has demonstrated antitumor activity among various solid tumor cell lines. Siemeister et al showed in a preclinical study that roniciclib strongly improved the efficacy of the standard-of-care combination treatment with the DNA-damaging agents cisplatin and etoposide in the Rb-negative NCI-H82 SCLC xenograft model without further worsening the tolerability of the therapy.33Subsequently, the randomized, phase II CONCEPT- SCLC trial was conducted to investigate the safety, efficacy, and clinical benefit of roniciclib versus placebo, when given in combination with cisplatin/carbopla-
References:
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi: 10.1016/j.cell.2011.02.013.
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6(4):353-367. doi: 10.1158/2159-8290.CD-15-0894.
- Ramanathan Y, Rajpara SM, Reza SM, et al. Three RNA polymerase II carboxyl- terminal domain kinases display distinct substrate preferences. J Biol Chem.
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing speci c inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704-707.
- Serrano M, Gómez-Lahoz E, DePinho RA, Beach D, Bar-Saqi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267(5195):249-252.
- Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264(5157):436-440.
- Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68;67-108.
- Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672-1677.
- Fry DW, Harvey PJ, Keller PR, et al. Speci c inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427-1438.
- Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as rst-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25-35. doi: 10.1016/S1470-2045(14)71159-3.
- Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): nal analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425-439. doi: 10.1016/S1470-2045(15)00613-0.
- Dickson MA, Tap WD, Keohan ML, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-ampli ed well-di erentiated or dedi erentiated liposarcoma. J Clin Oncol. 2013;31(16):2024-2028. doi: 10.1200/ JCO.2012.46.5476.
- Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as rst-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738-1748.
- Gazzeri S, Gouyer V, Vour’ch C, Brambilla C, Brambilla E. Mechanisms of p16INK4A inactivation in non-small-cell lung cancers. Oncogene. 1998;16(4):497-504.
- Konstantinidou G, Ramadori G, Torti F, et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013;3(4):444-457. doi: 10.1158/2159-8290.CD-12-0388.
- Schuster K, Venkateswaran N, Rabellino A, Girard L, Peña-Llopis S, Scaglioni PP. Nullifying the CDKN2AB locus promotes mutant K-ras lung tumorigenesis. Mol Cancer Res. 20149;12(6):912-923. doi: 10.1158/1541-7786.MCR-13-0620-T.
- Tam KW, Zhang W, Soh J, et al. CDKN2A/p16 inactivation mechanisms and their relationship to smoke exposure and molecular features in non- small-cell lung cancer. J Thorac Oncol. 2013;8(11):1378-1388. doi: 10.1097/ JTO.0b013e3182a46c0c.
- Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15(24):3249-3262.
- Puyol M, Martín A, Dubus P, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63-73. doi: 10.1016/j.ccr.2010.05.025.
- PatnaikA,RosenLS,TolaneySM,etal.E cacyandsafetyofabemaciclib,an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6(7):740-753. doi: 10.1158/2159-8290.CD-16-0095.
- Tao Z, Le Blanc JM, Wang C, et al. Coadministration of trametinib and palbociclib radiosensitizes KRAS-mutant non-small cell lung cancers in vitro and in vivo. Clin Cancer Res. 2016;22(1):122-133. doi: 10.1158/1078-0432.CCR-15-0589.
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519-525. doi: 10.1038/nature11404.
- Gopalan PK, Pinder MC, Chiappori A, et al. A phase II clinical trial of the CDK 4/6 inhibitor palbociclib (PD 0332991) in previously treated, advanced non-small cell lung cancer (NSCLC) patients with inactivated CDKN2A [ASCO abstract 8077]. J Clin Oncol. 2014;32(suppl):S5.
- Stephenson JJ, Nemunaitis J, Joy AA, et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2014;83(2):219-223. doi: 10.1016/j.lungcan.2013.11.020.
- Parry D, Guzi T, Shanahan F, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9(8):2344-2353. doi: 10.1158/1535-7163.MCT-10-0324.
- Mita MM, Mita AC, Moseley J, et al. A phase I study of the CDK inhibitor dinaciclib (SCH 727965) administered every 3 weeks in patients (pts) with advanced malignancies: Final results [ASCO abstract 3080]. J Clin Oncol. 2011;29(suppl).
- Bates DJ, Salerni BL, Lowrey CH, Eastman A. Vinblastine sensitizes leukemia cells to cyclin-dependent kinase inhibitors, inducing acute cell cycle phase- independent apoptosis. Cancer Biol Ther. 2011;12(4):314-325.
- Booher R, Hatch H, Dolinski BM, et al. MCL1 and BCL-xL levels in solid tumors are predictive of dinaciclib-induced apoptosis. PLoS One. 2014;9(10):e108371. doi: 10.1371/journal.pone.0108371.
- Li J, Xu M, Yang Z, Li A, Dong J. Simultaneous inhibition of MEK and CDK4 leads to potent apoptosis in human melanoma cells. Cancer Invest. 2010;28(4):350-356. doi: 10.3109/07357900903286966.
- Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512-6525.
- Wood A, Krytska K, Ryles HT, et al. Dual ALK and CDK4/6 inhibition demonstrates on-target synergy against neuroblastoma. Clin Cancer Res. 2016. pii: clincanres.1114.2016.
- Wikman H, Kettunen E. Regulation of the G1/S phase of the cell cycle and alterations in the RB pathway in human lung cancer. Expert Rev Anticancer Ther. 2006;6(4):515-530.
- Siemeister G, Lücking U, Wengner AM, et al. BAY 1000394, a novel cyclin- dependent kinase inhibitor, with potent antitumor activity in mono- and in combination treatment upon oral application. Mol Cancer Ther. 2012;11(10):2265- 2273. doi: 10.1158/1535-7163.MCT-12-0286.
- Reck M, Horn L, Novello S, et al. Phase II study of roniciclib in combination with cisplatin/etoposide or carboplatin/etoposide as rst-line therapy in subjects with extensive-disease small cell lung cancer (ED-SCLC). Ann Oncol. 2016;27 (Suppl 6):vi493-496. doi: 10.1093/annonc/mdw389.4.
- Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476-487. doi: 10.1093/jnci/djs002.
- Roberts PJ, White HS, Sorrentino JA, et al. Evaluation of targeted bone marrow arrest by G1T28, a CDK4/6 inhibitor in clinical development to reduce chemotherapy- induced myelosuppression [ASCO abstract 2529]. J Clin Oncol. 2015;33(suppl).













Ornstein Advises on Starting Dose and Management of Lenvatinib in RCC
April 21st 2024During a Case-Based Roundtable® event, Moshe Ornstein, MD, MA, provided guidance on dosing and toxicity concerns in a patient treated with lenvatinib plus pembrolizumab for advanced renal cell carcinoma.
Read More

