Chimeric Antigen Receptor T-Cell Therapy in Acute Myeloid Leukemia
Although preclinical data are promising, there are many clinical challenges in developing CAR T-cell therapy in acute myeloid leukemia.
Amanda Przespolewski, DO
Abstract
Chimeric antigen receptor (CAR) T-cell therapy is a novel form of cellular immunotherapy in which autologous T cells are genetically modified to express CARs found on tumor cells, as well as on costimulatory molecules required for T-cell activation. Although now an established treatment modality for CD19+ lymphoid malignancies, the development of CAR T-cell strategies for myeloid malignancies, specifically acute myeloid leukemia (AML), is still being explored preclinically and in phase I trials. Barriers to successful adaptation of this approach in AML include heterogeneous target antigen expression on diverse myeloid cell populations, co-expression on normal hematopoietic stem cells leading to off-target toxicity, and lack of clear anti-leukemia responses. This review will discuss the current status of CAR T-cell therapy in AML, highlighting the challenges and potential for this approach to change the treatment landscape for this disease.
Introduction
Adoptive immunotherapy holds much promise for the therapy of hematologic malignancies, with CD19+ lymphoid malignancies being the showpiece for this therapy. In chimeric antigen receptor (CAR) T-cell therapy, autologous T cells are genetically engineered to express CARs found on tumor cells as well as on costimulatory molecules.1Multiple early-phase trials investigating CAR T-cell therapy in treatment-refractory lymphoid malignancies, including chronic lymphocytic leukemia and acute lymphoblastic leukemia (ALL), have demonstrated significant antitumor effects, with high complete remission rates and clear reduction in tumor burden.2Such studies have sparked interest in the development of CAR T-cell therapy for myeloid malignancies, specifically acute myeloid leukemia (AML). However, major challenges in developing targeted immunotherapeutic approaches for AML, as compared with its lymphoid counterparts, are the underlying biological heterogeneity of myeloid malignancies and the lack of truly tumor-specific surface antigens that are also not expressed on normal myeloid and hematopoietic progenitor cells. To date, investigators have explored targeting CD33, CD123, and folate receptor (FR) beta as well as numerous other antigens as CAR T-cell constructs in preclinical models and limited early phase testing.3Here, we review the current status of CAR T-cell therapy for treatment of AML, highlighting the early
stages of development of this approach, clinical challenges, and its promise to change the treatment paradigm for this disease.
Preclinical Studies
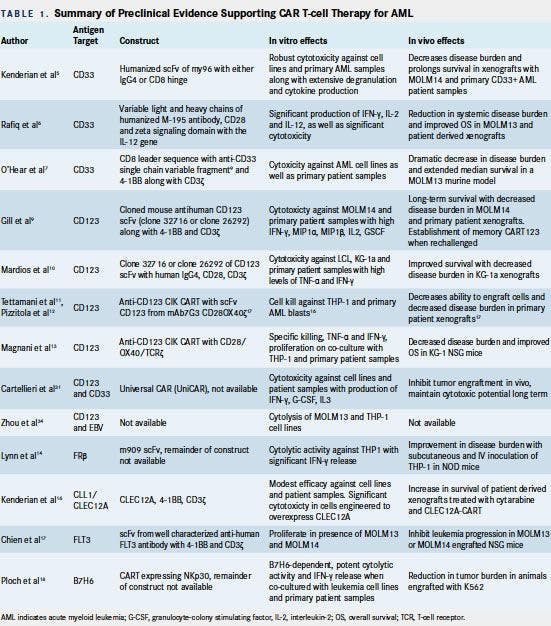
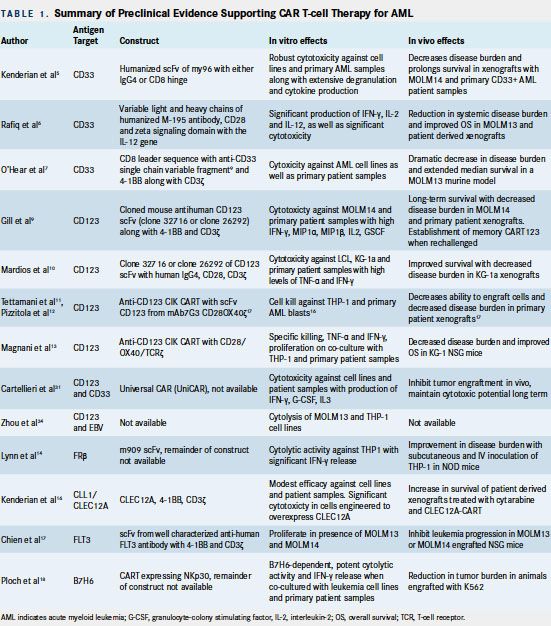
Significant preclinical data supporting the efficacy of CAR T-cell therapy for AML are summarized in Table 1.
CD33 Targeting
CD33 is a sialic acidbinding transmembrane receptor expressed on AML blasts, early multilineage hematopoietic progenitors, and myelomonocytic precursors, as well as, to a lesser degree, pluripotent stem cells and nonhematologic tissues.3The impetus for selection of the CD33 moiety as a target for CAR T-cell therapy arose from the known clinical activity of gemtuzumab ozogamicin, an anti-CD33 monoclonal antibodydrug conjugate, for treatment of CD33+ relapsed/refractory (r/r) patients with AML.4Kenderian et al designed CD33-targeted CAR T cells (CART-33), expressing a humanized version of the same single-chain variable fragment (scFv) of the original murine clone my96 along with either human CD8 or immunoglobulin G (igG)4 hinges.5Coculture of CART-33 with AML cells in vitro induced extensive CART-33 proliferation, degranulation, and cytokine production, as well as robust cytotoxicity at low effector-to-target ratios. Significant in vitro activity was observed against primary AML cells, with superior cytotoxicity seen with the CD4 hinge construct. Infusion of 1 x 105 CART-33 cells eradicated in vivo disease and prolonged overall survival (OS) of multiple primary CD33+ AML xenograft models (Table 1). Rafiq and colleagues generated CART-33 expressing variable light and heavy chains of the humanized M-195 antibody together with the costimulatory CD28 and CD3-zeta signaling domains with the interleukin (IL)-12 gene.6Co-culture of these T cells with AML cells resulted in enhanced interferon-γ (IFN-γ), IL-2, and IL-12 production. Significant cytotoxicity was observed in vitro in a range of effector-to-target ratios using AML cell lines and patient-derived CD33+ AML xenografts. O’Hear et al engineered alternative CART- 33 cells consisting of a CD33 single-chain variable fragment linked to a CD8 leader sequence, and the cytoplasmic domains of 4-1BB and CD3ζ. These cells exhibited cytotoxicity against a panel of 6 human CD33+ AML cell lines as well as patient samples. Infusion of these CART-33 cells in a murine model reduced leukemia burden to <1% of control animals.7
CD123 Targeting
CD123, a transmembrane α subunit of the IL-3 receptor, represents an attractive immunotherapeutic target for AML given its preferential overexpression in leukemic stem cells relative to normal CD34+ cells.8Gill et al generated CD123-directed CAR T cells (CART-123) using a construct comprised of cloned light-to-heavy or heavy-to-light orientations of the mouse anti-human CD123 scFv (clone 32716 or clone 26292) together with 41BB and CD3-zeta.9These CART-123 cells demonstrated significant cytotoxicity against AML cells and primary patient samples, and they induced IFN-gamma, macrophage inflammatory protein (MIP) 1α, MIP1β, IL-2, and granulocyte macrophage colony-stimulating factor (G-CSF). In vivo, this construct led to decreased disease burden in CD123+ AML cell lines and primary patient xenografts. Of note, animals rechallenged with MOLM14 AML cells following previous CART-123 therapy exhibited rapid proliferation of CART-123 cells consistent with establishment of T-cell memory. Mardios et al generated a similar CART-123 construct comprised of human igG4 Fc region, CD28, and CD3-zeta signaling domains.10These CART-123 cells produced high levels of tumor necrosis factor (TNF)-alpha and IFN-gamma, and they demonstrated both cytotoxic activity and proliferation in the presence of CD123+ AML cell lines and patient samples but not in the presence of CD123cells. Decreased disease burden and improved OS were observed in xenograft models. Cytokine-induced killer (CIK) cells possessing spontaneous antitumor activity have also been transduced to express an anti-CD123 CAR via an SFG retroviral vector, also known as a Moloney murine leukemia virus-based plasmid, utilized to transfect the cells of interest.11These cells have the advantage of displaying nonhuman leukocyte antigen-restricted cytotoxicity and minimal alloreactivity. Anti-CD123 CIK CART-cells exhibited high (60%) in vitro cell kill against CD123+ THP-1 cells and primary AML blasts. An alternative CART engineered with ScFv-CD123 generated from the mAb 7G3 and CD28/OX40-zeta constructs also demonstrated decreased tumor burden in primary AML patient xenografts.12Magnani and colleagues showed the feasibility and efficacy of a nonviral methodology (the Sleeping Beauty transposon system) to produce CAR CIKs.13 CD123 CIK cells generated with this approach, with CD28/OX40/TCRζ costimulatory domains, demonstrated competent in vitro T-cell responses and decreased in vivo disease burden in mice following infusion of 1 x 107 CAR CIK cells every 10 days.

Folate Receptor β Targeting FRβ is a cell membranebound folate receptor with homology with folate receptor alpha, a well established target for CAR T cells in epithelial tumors. FR-beta is expressed in 70% of primary AML cases, with relatively limited expression in normal tissue. Using m909 scFv, FR-beta-specific CAR constructs were generated, which were shown to have significant
IFN-gamma release response to FR-beta+ AML targets compared with CD19-directed CAR T-cell controls.14The m909 CAR T cells exhibited proliferation and lysis when co-cultured with AML cells with high to moderate FR-beta expression. Further, m909 CAR T cells demonstrated in vivo antitumor activity.
Human C-Type Lectin-Like Molecule-1 Targeting
Human C-type lectin-like molecule-1 (CLL1 or CLEC12A) is another cell surface marker overexpressed in AML blasts and leukemic stem cells. Because AML blasts express CLL1 heterogeneously, CAR T cells engineered with this antigen show only modest effector
functions against primary AML cells. However, because CLL1 is preferentially overexpressed in AML cells in the marrow of patients who fail to achieve complete remission after induction chemotherapy, these CAR T-CLL1 cells may represent an effective means of eradicating minimal residual disease (MRD). Mice engrafted with primary human AML who received cytarabine followed by a single dose of CAR T-CLL1 lived significantly longer relative to untransduced mice receiving only chemotherapy (100% survival at 200 days compared with 20% in controls).15,16
FLT3 Targeting
FMS-like tyrosine kinase-3 (FLT3) is expressed on the surface of normal hematopoietic progenitors as well as on most AML blasts, independent of FLT3 mutation status, leading to active investigation of FLT3 as a target for CART-directed AML therapy. T cells
have been transduced with a FLT3 CAR construct consisting of an scFv derived from a well-characterized antihuman FLT3 antibody paired with 41BB and CD3ζ costimulatory domains. These CART-FLT3 cells demonstrated in vitro proliferation when exposed to FLT3+ AML cell lines, and they inhibited leukemia progression in nonobese diabetic scid gamma mice engrafted with the same cells. Of note, comparison of CART-FLT3 with CART-33 cells revealed that CARTFLT3transduced cells appear to have equivalent or potentially less toxicity to normal hematopoietic progenitors than do their CD33 counterparts, suggesting a potential advantage to this targeted approach.17
B7H6 Targeting
B7H6, a member of the B7 family, is readily detectable on AML blasts but not on healthy tissue, and it is recognized by NKp30, an activating receptor on NK cells. CAR T cells expressing NKp30 elicited B7H6-dependent potent cytolytic activity and IFN-gamma release when cocultured with various leukemia cell lines and primary blasts, but not with B7H6 myeloma lines. AML engrafted mice infused with NKp30+ CAR T cells exhibited appreciable reduction in tumor burden. Treatment was particular effective in vivo in eradicating minimal residual AML disease (defined as 5% blasts).18
Lewis Y Targeting
Lewis Y (LeY) antigen is also overexpressed on AML cells, with limited expression on normal tissues. Preclinical studies have demonstrated that LeYspecific CAR T cells produced varying amounts of IFN-gamma on exposure to AML cell lines and primary AML cells. In addition, co-culture of the Lewis Y CAR T-cell construct with these cell lines resulted in cytolytic activity.19
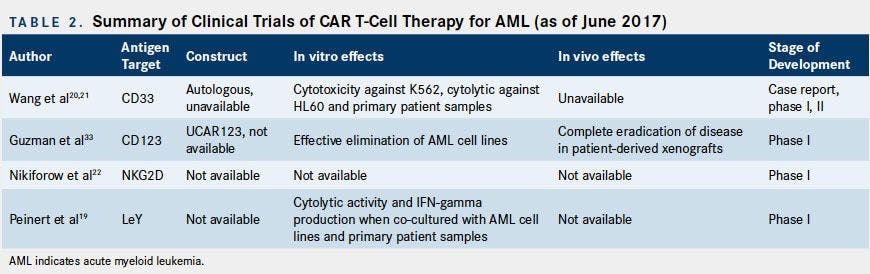
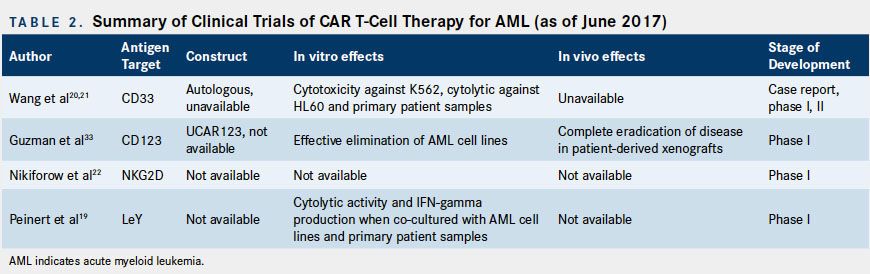
Clinical Studies
Summarized below and in Table 2 are the available limited published clinical data on CAR T-cell therapy for AML.
CART-33 Clinical Studies
In the only published case report, Wang and colleagues reported the efficacy and tolerability of autologous CART-33 for a single patient with refractory AML.20A total of 1.12 x 109 CART-33 cells were administered in escalating doses over 4 consecutive days
without conditioning therapy. Following infusion, the patient experienced a marked decrease in blast ratio from >50% pre-treatment to <6% at 2 weeks following administration. Despite persistently high levels of CAR T cells as induction of multiple immunomodulatory
cytokines (IL-6, IL-8, TNF-alpha, and IFN-gamma) in the peripheral blood, the blast percentage gradually increased to nearly 70% at 9 weeks post infusion, suggesting tumor escape mechanism. The patient ultimately succumbed to his disease at 13 weeks post CAR T-cell infusion. Toxicities included grade 4 chills, fevers with T-max 42°C treated with etanercept (antiTNF-alpha) and transient hyperbilirubinemia. Two ongoing clinical trials in China are currently underway involving CART-33 cells for patients with AML.21
NCT01864902 is a continuation of the phase I study by Wang and colleagues; its goal is to evaluate the safety and in vivo survival of autologous or donor-derived CART-33 cells in patients with relapsed/refractgory (r/r) AML. The second trial (NCT02799680) is a phase
I study evaluating the safety and efficacy of allogeneic CART-33 cells in patients with r/r AML.
CART-123 Clinical Studies
Multiple trials for CART-123 T cells in AML are underway. A phase I trial for UCAR123 is currently enrolling patients with r/r AML (NCT03190278).21Another phase I dose escalation study is investigating the safety, tolerability, and optimal dosing of anti CD123/CD28 CAR T cells that also express truncated EGFR. Patients enrolled in this study will undergo lymphodepletion with cyclophosphamide, fludarabine, or etoposide, followed by T-cell infusion (NCT02159495). Lastly, a phase I trial evaluating donor-derived CART-123 cells in relapsed AML following allogeneic transplant is currently recruiting participants (NCT03114670).

NKG2D Ligand CAR T Cells
NKG2D ligands are upregulated on the cell surface of malignant hematopoietic cells, and thus they may serve as another CAR T cell targeting candidate. Nikiforow and colleagues recently conducted a feasibility and safety trial of CAR T cells targeting NKG2D in 6 patients with AML/myelodysplastic syndromes. Single doses of CAR T cells were well tolerated with no dose-limiting toxicities or cytokine release. Following single dosing, no patients had appreciable tumor response 28 days after infusion. Future studies investigating the effects of multiple CAR T-cell infusions are currently underway.22
Lewis Y CAR T-Cells
Anti-LeY CAR T cells were evaluated in a phase I trial of relapsed AML at Peter MacCallum Cancer Centre in Australia (NCT01716364). In this study, patients received fludarabine and cytarabine followed by CAR T-cell infusion. Out of 4 evaluable patients, 1 achieved a transient cytogenetic remission (5 months), 1 experienced a transient reduction in peripheral blood blasts, and 1 had stable disease for 23 months. No grade 3 or 4 toxicities or cytokine release syndrome were observed. Although all patients eventually relapsed, anti-LeY CAR T cells persisted for up to 10 months.23
Challenges and Future Directions
Despite the dramatic effects of CAR T-cell therapy on preclinical models, numerous clinical challenges remain as barriers to the successful implementation of this novel immunotherapeutic approach for treatment of myeloid malignancies in patients. These include: a) heterogeneous antigen expression on diverse AML cell populations; b) potential for off-target toxicities to normal myeloid progenitor and hematopoietic stem cells in patients; and c) potential for life-threatening complications of potent T-cell activation in vivo, specifically cytokine release syndrome and fatal neurological events. To date, much emphasis has been placed on finding optimal antigenic targets, minimizing off-tumor effects including cytokine release syndrome, and abrogation of unchecked cytotoxic T-cell (CTL) activity
resulting in myeloablative hematological toxicity.
Given the heterogeneous expression of CD33, CD123, and FRβ across AML cell profiles, there is vested interest in broadening the targeting milieu of CAR T-cell therapies. Ongoing studies by Perna and colleagues are aimed at identifying novel CAR antigenic targets for AML therapy.24Probing of the AML surfaceome for candidate molecules, followed by flow cytometric validation analyses, recently resulted in 4 novel antigen candidates, including 2 G-protein coupled receptors not previously reported. In pairing candidate antigens, validation studies have shown 3 distinct antigen combinations that are ~100% expressed on AML cells but are absent on normal cells.
Multiple safety mechanisms are also currently being incorporated into CAR T-cell technology to address concerns regarding potential off-target toxicity and the CTL activity on normal myeloid and hematopoietic progenitors. For instance, CART-33 therapy reduces CD34+ CD38 hematopoietic stem cells as well as CD34+ CD38– myeloid progenitors and peripheral myeloid cells in mice engrafted with human fetal CD34+ cells.5In addition, of concern, significant ablation of normal hematopoiesis has also been observed following CART-123 infusions.9Suicide gene systems as a means of mitigating damage to normal cells during adoptive cell therapies25,26have previously been shown to be efficacious in controlling graft versus host disease (GVHD) in allogeneic hematopoietic stem cell transplant recipients. This technology is currently being adapted to engineer CAR T cells.27CART-33 cells with available activating elimination gene components to truncate T-cell activity have also been designed.6,28Genetically engineered systems using inducible caspase-9mediated apoptosis pathways have also been explored to abrogate CAR T-cell toxicity against normal hematopoietic progenitors.29The GoCAR-T platform, combining a CD123-specific CAR construct with a costimulatory switch, has recently developed.30
To improve efficacy in targeting heterogeneous myeloid malignancies, dual targeting of CD123 and CD33 utilizing the universal CAR (UniCAR) model has recently emerged as an innovative approach.31This modality provides added flexibility to antigen selection strategies by allowing for the introduction of multiple targeting modules (TMs) either simultaneously or in sequence during treatment. During in vitro evaluation utilizing this model, dual-specific anti-CD123-CD33 TMs showed enhanced anti-leukemic effects, as evidenced by the low concentrations of bispecific TMs needed to elicit target-cell lysis. In subsequent studies, UniCAR cells interacting with dual-specific TMs effectively eradicated >80% blast cells in primary AML samples. T-cell specific cytokines (IFN-gamma, G-CSF, IL-13) were also detected following treatment, indicating viable UniCAR activation in response to bispecific TMs. By targeting multiple antigenic variants either simultaneously or sequentially, it is also feasible to reduce the emergence of antigen-loss tumor escape mechanisms. The same investigators recently demonstrated that UniCAR cells exhibit superior antitumor reactivity to conventional CAR T cells at low effector-to-target ratios.32Infusion of cells engineered to express CD123-specific target modules resulted in a) inhibition of AML engraftment in murine hosts, and b) long-term persistence. UniCAR cells obtained from said mice retained cytotoxic potential against AML cell lines for at least 4 months after transplantation, supporting the potential for long-term immunoprotection.
The use of UniCAR/UniTARG (UniTARG is an antibody-based platform with universal effector arm with individual targeting molecules), a flexible, modular CAR platform, also represents an alternative means of modulating off-tumor, on-target effects with CART therapy.31,32Conventional, second-generation CAR signaling is contingent on tumor surface antigen recognition by scFv expressed on retargeted T cells. Within the UniCAR paradigm, there is a separation of the antigen-binding and antigen-signaling aspects of the system. Engineered cells express a UniCAR that has specificity for a short peptide motif derived from human nuclear protein rather than a specific tumor antigen. Such cells remain physiologically inert after reinfusion until specific TMs carrying both tumor specific antigen and the nuclear peptide motif are introduced. Guzman and colleagues have taken this approach 1 step further by engineering CD123-directed T cells that no longer express T-cell receptor (TCR); this was done as a means to avoid the potentially life-threatening clinical effects of potent T-cell activation (ie, cytokine release syndrome, fatal cerebral edema) that have been reported in CART-19 T-cell therapy in patients with ALL.33These specifically modified UCAR123 cells have minimal to no risk of inducing GVHD in recipients, but they retain anti-leukemic activity as demonstrated by effective in vitro elimination of AML cells and complete AML eradication in the marrow of patient-derived xenografts. Mice treated with these modified UCAR123 cells also showed significant increases in OS as compared with control mice.
Furthermore, there has been interest in developing multiple targeted CAR T cells against both tumor and nontumor surface antigens. Given the high mortality and morbidity associated with viral infections following intensive chemotherapy and allogeneic stem cell transplant for acute leukemias, Zhou et al recently engineered CD123-directed CAR T cells that also recognize virus-specific epitopes of Epstein-Barr virus (EBV), adenovirus, and cytomegalovirus.34Both EBV-specific and multi-virusspecific CAR T cells demonstrate virus-specific cytotoxicity against EBV and mediate AML cell lysis. This approach may ultimately yield a 1-stop multidimensional treatment option for AML and unresolved common viral infections in this patient population.
Recent studies have investigated the possibility of combining CAR T cells with checkpoint inhibitors to further enhance antileukemic activity. Emerging data in many disease types have shown that exposure of malignant cells to directed CAR T cells induces expression of the programmed death (PD) ligands 1 and 2 on tumor cells. Subsequent upregulation of the receptor for PD-1 on CAR T cells constitutes an exhausted T-cell phenotype. Kenderian et al demonstrated that in vitro co-culture of human AML cells (primary AML patient samples and the AML cell line MOLM14) with AML-directed CAR T cells enhanced expression of multiple coregulatory molecules (PD-1 and TIM-3) on the T cells.35Exposure of CAR T cells harvested from AML-bearing murine hosts to PD-1 or TIM-3 blockade effectively prevented immune exhaustion and resulted in increased CAR T-cell proliferation and cytokine production. In vivo administration of PD-1 or TIM-3 blockade with CAR T-cell therapy translated into improved disease control and OS in leukemia mouse models. This combination warrants further clinical investigation in future clinical trials.
Lastly, another promising cellular immunotherapeutic approach for AML is NK cell therapy. Patients with AML exhibit impaired NK cell function. Allogeneic NK cell infusions in nontransplanted and transplanted patients with AML have been shown to mediate anti-leukemic effects without inducing graftversus-host activity. Sources of NK cell infusions include alloreactive or haploidentical cells, as well as NK cells derived from normal hematopoietic progenitor cells in umbilical cord blood or bone marrow, and G-CSF-mobilized peripheral stem cells. The ability to generate high numbers of NK cells ex vivo from CD34+ hematopoietic precursors may allow this technology to serve as a universal “off the shelf” cellular therapy for most patients with AML. To date, adoptive NK cell therapy has shown promising activity preclinically and in early-phase clinical trials in limited numbers of patients. NK cell therapy may be particularly promising for the treatment of patients with chemoresistant disease as a bridge to transplant or for treating patients in the MRD state. Ongoing clinical investigations are addressing optimal cell dosage, toxicities, duration of NK cell persistence, and how best to maintain the in vivo activation status of infused NK cells, ie through concomitant cytokine support (IL-2, IL-15).36, 37
Conclusion
Emerging rigorous preclinical data have paved the way for the further clinical development of CAR T-cell therapy for AML. However, many clinical challenges remain. These include appropriate target selection, lack of potent anti-leukemic effects, and risks of myeloablative and cytolytic T-cell toxicities. At present, both a) the identification of new targetable antigens, and b) the development of a flexible modular CAR platform capable of dual or sequential targeting of antigens, hold promise to address these issues. The results of ongoing clinical trials testing the safety of numerous CAR T-cell constructs in patients with AML are eagerly awaited. Once completed, further studies of CAR T cells for treatment of overt AML disease, as well as for MRD following chemotherapy and/or transplantation, should be explored.
Acknowledgments
This work was supported by Roswell Park Cancer Institute (RPCI) and by National Cancer Institute grant P30CA016056. EW is also supported by the RPCI Alliance Foundation (Jacquie Hirsch Leukemia Research Fund).
References:
- Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13(8):525-541. doi: 10.1038/nrc3565.
- Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312-3320. doi: 10.1182/blood-2016-02-629063.
- Dinndorf PA, Andrews RG, Benjamin D, et al. Expression of normal myeloidassociated antigens by acute leukemia cells. Blood. 1986;67(4):1048-1053.
- Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369-377. doi: 10.1200/JCO.2010.31.4310.
- Kenderian SS, Ruella M, Shestova O, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637-1647. doi: 10.1038/leu.2015.52.
- Rafiq S, Purdon TJ, Schultz LM, Brentjens RJ. CD33-directed chimeric antigen receptor (CAR) T cells for the treatment of acute myeloid leukemia (AML). Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- O’Hear C, Heiber JF, Schubert I, et al. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. 2015;100(3):336-344. doi: 10.3324/haematol.2014.112748.
- Testa U, Riccioni R, Diverio D, et al. Interleukin-3 receptor in acute leukemia. Leukemia. 2004;18(2):219-226.
- Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014; 123(15):2343-54.
- Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123- specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138-3148. doi: 10.1182/blood-2012-474056.
- Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389-401. doi: 10.1111/ bjh.12282.
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28(8):1596-1605. doi: 10.1038/leu.2014.62.
- Magnani CF, Turazzi N, Benedicenti F, et al. Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform. Oncotarget. 2016;7(32):51581-51597. doi: 10.18632/oncotarget.9955.
- Lynn RC, Poussin M, Kalota A, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125(22):3466-3476.
- Lu H, Zhou Q, Deshmukh V, et al. Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew Chem Int Ed Engl. 2014;53(37):9841-9845. doi: 10.1002/anie.201405353.
- Kenderian SS, Ruella M, Shestova O, et al. Leukemia stem cells are characterized by CLEC12A expression and chemotherapy refractoriness that can be overcome by targeting with chimeric antigen receptor T cells. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Chien CD, Sauter CT, Ishii K, et al. Preclinical development of FLT3-redirected chimeric antigen receptor T cell Immunotherapy for acute myeloid leukemia. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Ploch P, Khan S, Bhatti A, et al. Exploring chimeric natural killer receptor NKP30 expressing human T lymphocytes for adoptive immunotherapy to acute leukemia. Presented at: 21st Congress of the European Hematology Association; June 9-12, 2016; Copenhagen, Denmark. https://learningcenter. ehaweb.org/eha/2016/21st/133348/udo.hartwig.exploring.chimeric. natural.killer.receptor.nkp30.expressing.human.html?f=m1.
- Peinert S, Prince HM, Guru PM, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther. 2010;17(5):678-686. doi: 10.1038/gt.2010.21.
- Wang QS, Wang Y, Lv HY, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184-191. doi: 10.1038/mt.2014.164.
- Study Evaluating Safety and Efficacy of UCART123 in Patients with Acute Myeloid Leukemia. clinicaltrials.gov/ct2/show/NCT03190278?term=cd123+ car&cond=Acute+Myeloid+Leukemia&rank=4. Updated September 13, 2017. Accessed June 20, 2017.
- Nikiforow S, Werner L, Murad J, et al. Safety data from a first-in-human phase 1 trial of NKG2D chimeric antigen receptor-T cells in AML/MDS and multiple myeloma. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122-2129. doi: 10.1038/mt.2013.154.
- Perna F, Berman S, Mansilla-Soto J, et al. Probing the AML surfaceome for chimeric antigen receptor (CAR) targets. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Minagawa K, Zhou X, Mineishi S, Di Stasi A. Seatbelts in CAR therapy: how safe are CARS? Pharmaceuticals (Basel). 2015;8(2):230-249. doi: 10.3390/ ph8020230.
- Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. doi: 10.3389/ fphar.2014.00235.
- Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673-1683. doi: 10.1056/ NEJMoa1106152.
- Song D, Swartz MH, Biesecker SG, et al. Chimeric antigen receptor-modified T cells for the treatment of acute myeloid leukemia expressing CD33. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Thokala R, Olivares S, Mi T, et al. Redirecting specificity of T cells using the Sleeping Beauty system to express chimeric antigen receptors by mixand- matching of VL and VH domains targeting CD123+ tumors. PLoS One. 2016;11(8):e0159477. doi: 10.1371/journal.pone.0159477.
- Foster AE, Duong ML, Lu A, et al. Inducible MyD88/CD40 (iMC) costimulation provides ligand-dependent tumor eradication by CD123-specific chimeric antigen receptor T cells. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Cartellieri M, Feldmann A, Koristka S, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(8):e458. doi: 10.1038/bcj.2016.61.
- Loff S, Bonin MV, Dietrich J, et al. The UniCAR system: inducible CAR T cells for precise reactivity and high efficacy against hematopoietic malignancies [abstract]. In: Proceedings of the Second CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference: Translating Science into Survival. Cancer Immunology Research. 2016:B099-B099. doi: 10.1158/2326-IMM2016-B099.
- Guzman ML, Sugita M, Zong H, et al. Allogeneic Tcrα/β deficient CAR T-cells targeting CD123 prolong overall survival of AML patient-derived xenografts. Presented at: American Society of Hematology 58th Annual Meeting & Exposition; December 3-6, 2016; San Diego, CA.
- Zhou L, Liu X, Wang X, et al. CD123 redirected multiple virus-specific T cells for acute myeloid leukemia. Leuk Res. 2016;41:76-84. doi: 10.1016/j. leukres.2015.12.003.
- Kenderian SS, Ruella M, Shestova O, et al. Identification of PD1 and TIM3 as checkpoints that limit chimeric antigen receptor T cell efficacy in leukemia. Blood. 2015;126:852.
- Sinha C, Cunningham LC. An overview of the potential strategies for NK cell-based immunotherapy for acute myeloid leukemia. Pediatr Blood Cancer. 2016;63(12):2078-2085. doi: 10.1002/pbc.26171.
- Dolstra H, Roeven MWH, Spanholtz J, et al. Successful transfer of umbilical cord blood CD34+ hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res. 2017;23(15):4107- 4118. doi: 10.1158/1078-0432.CCR-16-2981.











Peers Discuss Role of Pola-R-CHP vs R-CHOP in Newly Diagnosed DLBCL
April 19th 2024During a Case-Based Roundtable® event, Haifaa Abdulhaq, MD discussed with participants whether the POLARIX trial influences their choice to use the pola-R-CHP as opposed to R-CHOP regimen for patients with newly diagnosed diffuse large B-cell lymphoma.
Read More


Powell Reviews Updated IO/TKI Data and AE Management in Endometrial Cancer
April 18th 2024During a Case-Based Roundtable® event, Matthew A. Powell, MD, discussed the case of a patient with advanced endometrial cancer treated with lenvatinib plus pembrolizumab who experienced grade 2 treatment-related hypertension.
Read More


Savona Discusses First-Line JAK Inhibition for Patients With Myelofibrosis at Risk of Anemia
April 17th 2024During a Case-Based Roundtable® event, Michael Savona, MD, and participants discussed the case of a patient with myelofibrosis and moderate anemia receiving JAK inhibitor therapy.
Read More

