Overcoming Resistance to Immunotherapy Requires Advanced Strategies
Primary and acquired resistance to immunotherapy necessitates novel strategies that can overcome cellular processes and genetic mutations of resistance to advance into the next age of cancer therapy.
Michael A. Curran, PhD
Primary and acquired resistance to immunotherapy necessitates novel strategies that can overcome cellular processes and genetic mutations of resistance to advance into the next age of cancer therapy.
Despite the encouraging success of immune checkpoint inhibitors (ICIs) in recent years, roughly 60% to 70% of tumors are not responsive to single-agent ICI therapy, whereas those that do respond can become resistant over time.1
“Even in melanoma, which is considered one of the most immunotherapy-receptive tumors, 40% to 50% of patients do not respond to ICIs from day 1. After 3 years, the response rate is down to 35% to 40%, and the responses plateau from there,” said Michael A. Curran, PhD, assistant professor in the Department of Immunology at The University of Texas MD Anderson Cancer Center in Houston.
“Innate resistance is seen in 40% to 50% of patients, and another 20% to 30% of patients respond and relapse due to acquired resistance,” said Curran, who conducted his postdoctoral studies on the effect of checkpoint blockade on cancer progression under immunotherapy pioneer James P. Allison, PhD.
The current era of immuno-oncology was aptly punctuated this year when it was announced that Allison and Tasuku Honjo, MD, PhD, shared the 2018 Nobel Prize in Physiology or Medicine for their discoveries in cancer immunotherapy.
ICIs spawned directly from studies led by Allison and Honjo, among others, that exemplify the success of immunotherapy in subsets of cancers previously considered difficult to treat, such as advanced metastatic melanoma.
To date, ICIs have gained regulatory approval in melanoma, nonsmall cell lung cancer (NSCLC), renal cell carcinoma, and refractory bladder and head and neck cancers, among others.
In ongoing work, Curran and colleagues seek to identify the characteristics of the tumor microenvironment (TME) that drive resistance to immunotherapy in “immune-cold” cancers, which are nonresponsive to blockade of the immune checkpoint regulators CTLA-4 and the PD-1/PD-L1 system, along with strategies to sensitize them.
Julie R. Brahmer, MD
“Over half of all cancers are what we would consider immune-cold tumors, with an abysmal response rate to immunotherapyin the range of 5%,” Curran said. “If we look across all patients with all types of cancers, it is imperative to understand the sources of resistance to immunotherapy before we can achieve the goal of improving response rates and patient outcomes.”
The clinical response to ICIs has yet to be completely characterized; the current model for ICI-mediated antitu- mor response predicates activation and clonal proliferation of antigen-experienced T cells in the TME.2,3 The response involves many steps, including processing and presentation of tumor-associated antigens, recognition of tumor antigens displayed by major histocompatibility complex (MHC) via unique T-cell receptors (TCRs), activation and proliferation of CD8-positive tumor-specific T cells, and, ultimately, dif- ferentiation into T effector (Teff) cells and oncolysis.
Innate and acquired changes affecting any of these steps can impede antitumor immune responses driven by ICIs.4 Mecha- nistically, resistance and immunosuppression can stem from 2 sourcestumor-intrinsic genetic mutations and tumor- extrinsic local adaptive or systemic immunosuppression.
Julie R. Brahmer, MD, director of the Thoracic Oncology Program and professor of oncology at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Medicine in Baltimore, Maryland, noted that these mechanisms are not exclusive. “There are multiple routes to resistance, and patients may have 1 or, more likely, multiple mechanisms that result in resistance to ICIs,” she said in an interview with Targeted Therapies in Oncology.
Hostile Environment for Immune Mediators
Characteristics of the TME are emerging as dominant forces in promoting immunosuppression and resistance to immunotherapies. Curran noted that the effects of myeloid stroma and tumor hypermetabolism on reorganization of the TME are 2 emerging foci of what he calls a “miserable microenvironment”: Stromal and other immunosuppressive cells and metabolism-driven effects on TME collude to create a neighborhood hostile to infiltration, survival, proliferation, and oncolytic functions of antitumor T cells.
“T cells, for instance, are exquisitely sensitive to glucose levels and are unable to compete with tumor cells in this hostile environment,” Curran said. The rapid growth and proliferation of tumor cells results in a hypermetabolic state, with reliance of enhanced glycolysis and oxidative phosphorylation along with depletion of oxygen, glucose, and amino acids in the TME. Studies have shown that glucose depletion due to tumor glycolysis attenuates T-cell effector function and that tumor-expressed PD-L1 provides a constitutive “reverse signal” that promotes glycolysis.5,6Moreover, checkpoint blockade antibodies against CTLA-4, PD-1, and PD-L1 restored TME- glucose, potentiating T-cell glycolysis and interferon (IFN)-γ production, suggesting a role for checkpoint blockade in stabilizing tumor/TME metabolic balance.6
Tumor cells also reorganize the TME chemotactically to favor recruitment of suppressive myeloid stroma that favors growth, aggressiveness, and invasion of tumor cells, as well as excluding immune response mediators such as dendritic cells (DCs), which are necessary for antigen presentation.
The production of transforming growth factor (TGF)β is an example of an immunosuppressant abundant in the TME. Tumors, tumor-associated stromal cells, and regulatory T cells (Tregs) produce large quantities of TGF-β, a differentiation factor for myeloid and T cells that promotes myeloid-derived suppressor cell (MDSC) and Treg differentiation. Of note, many currently available, clinically applicable TGF-β antibodies have potential in alleviating TGF-β-mediated immunosuppression in the TME to restore ICI sensitivity.
Preclinical data also support a TGF-βtargeted strategy to sensitize tumors to immunotherapy. For instance, chimeric antigen receptor T cells engineered to target prostate-specific membrane antigen (PSMA) and be insensitive to TGF-β were able to kill prostate tumor cells specifically in a mouse model of metastatic castrate-resistant prostate cancer.7
The Role of Hypoxic Stress in Immunotherapy Resistance
Another consequence of tumor hypermetabolism is that the rapid angiogenesis and oxygen consumption render the TME acidic and hypoxic.8 Hypoxic stress has a well-established role in cancer progression and therapeutic response; recently, regulatory mechanisms of hypoxia-mediated immunosuppression demonstrated how this common feature of all solid tumors drives resistance to immunotherapy and ICIs in particular.
Curran described hypoxia’s strong role in determining response to immunotherapy: “Prostate and pancreatic cancers, for instance, are very hypoxic and are also less responsive to ICI therapy than melanomas and lung cancers. For example, in the murine prostate cancer model we [use], about 40% of the tumor is hypoxic, meaning that nearly half the tumor is insensitive to immune checkpoint blockade.”
The acidity of the TME, combined with hypoxia, potently suppresses T-cell activation, proliferation, and cytotoxicity.8Lymphocytes undergo apoptotic cell death at the lower pH of the TME, whereas tumor cells thrive, for example. Hypoxia also fosters accumulation of extracellular adenosine, which inhibits proliferation and cytotoxicity of DCs, Teffs, and natural killer (NK) cells.
This adenosine accumulation is driven by hypoxia-inducible factor-1αmediated induction of the CD39 and CD73 ectonucleotidases and stimulating adenosine receptor (AR) expression. “Clinically available small molecule inhibitors of AR that are well tolerated may provide a strategy for counteracting the adenosine accumulation–dependent immunosuppression, complementing the effect of ICIs,” Curran added.


It is becoming evident that hypoxic stress in the TME affects nearly all steps of the antitumor cellular immune response. Hypoxia impairs T-cell infiltration, blunts the immune responses mounted by NK and NK-T cells, and attracts Tregs, tumor-associated macrophages, and MDSCs. It also contributes to intratumoral heterogeneity, thereby acting as a critical regulator of antitumor immunity and response/resistance to immunomodulatory therapies.
A deeper understanding of hypoxia-driven immunosuppression may be critical going forward for designing combination therapies that can help circumvent hypoxia-driven impediments to ICI and other immunotherapies. A recent study in an ICI-resistant murine model of prostate cancer exemplifies the potential and potency of targeting hypoxia in bolstering immunotherapy. In it, combination therapy with the hypoxia-activated prodrug TH-302 and checkpoint blockade synergized to cure over 80% of prostate tumors, a classically cold tumor.9
Despite the heterogeneity of hypoxia within and across tumor lesions, “the underlying processes that give rise to the hypoxia are fairly consistent across tumors,” Curran said. “We have been able to identify drugs across tumor types that can alleviate hypoxia and improve the ability of T [cells] to infiltrate and function.”
Undesirable Cellular Mediators of Immunosuppression
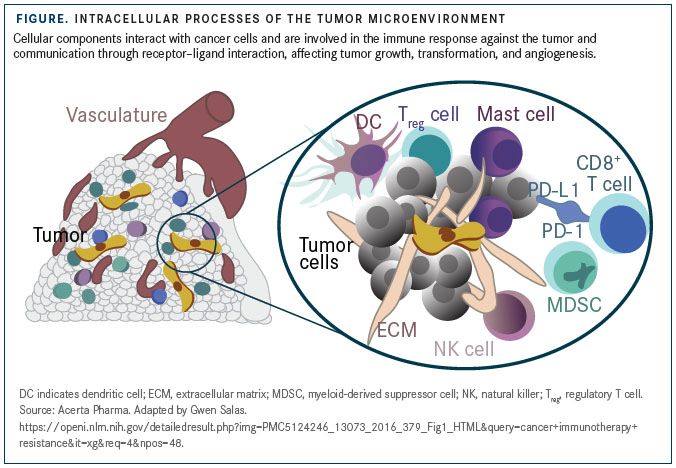
The infiltration of tumors by CD8-positive cytotoxic T cells, rather than inflammation-associated immune cells, is an indicator of favorable prognosis in several tumor types. DCs and NK cells, too, help mount antitumor responses by aiding tumor-antigen presentation and releasing perforin and granzyme B to induce oncolysis (FIGURE).
In counterpoint to this antitumor cellular activity, MDSCs and macrophages are major drivers of an immunosuppressive TME, as these cells deplete extracellular arginine, enhance tryptophan degradation, and sequester cystine in the TME.10For example, MDSCs have been shown to upregulate indoleamine 2,3-dioxygenase (IDO), an enzyme degrading L-tryptophan into N-formylkynurenine, resulting in T-cell arrest and anergy, and preclinical studies indicate that inhibition of IDO in combination with ICI therapy may improve response to checkpoint blockade.11,12
Tregs and cancer-associated fibroblasts (CAFs) are other TME components that contribute to immunosuppression and dampen response to ICIs. CAF-mediated immunosuppression is an elegant example of how these stromal residents in the TME subvert antitumor T-cell responses and promote “immune ignorance,” a state in which the immune system is unable to detect the presence of tumor cells.
CAFs were shown to sample, process, and cross-present antigen, killing CD8-positive T cells in an antigen-specific, antigen-dependent manner via PD-L2 and FAS ligand.13However, when T cells recognize the antigen and bind to these antigen-displaying CAFs, they undergo apoptosis instead of activation.
“These CAFs act as a decoy for CD8-positive T cells and draw them away from tumors and help kill tumor-infiltrating T cells before they can even detect or kill tumor cells,” Curran said. Thus, CAFs can support T-cell suppression in the TME by a mechanism dependent on immune checkpoint activation.
Mutation-Driven Resistance to ICIs
A significant number of proposed and now-characterized mechanisms of primary resistance have been defined, including low tumor mutational burden, which restrict the number of presented immunogenic antigens, nutrient depletion, secreted immunosuppressants, and immunosuppressive cells in the TME, as well as environmental (eg, gut microbiome), underlying genetic (eg, human leukocyte antigen type), and endocrine and metabolic (eg, chronic diseaseassociated immunosuppressive effects) factors. “In contrast, less is known about the mechanisms of acquired resistance,” Brahmer noted.
Mutation-driven desensitization to other classes of agents, such as tyrosine kinase inhibitors, are common features in acquired resistance. Analysis of the mutational landscape of tumors with acquired ICI resistance has yielded insights into some of the key players in this process.
An analysis of tumor neoantigens during the emergence of acquired resistance in patients with NSCLC after initial response to immune checkpoint blockade with single-agent antiPD-1 or in combination with anti–CTLA-4 found that genomic changes resulted in loss of nearly half the putative mutation-associated neoantigens in resistant clones.14
Other studies have reiterated the role of tumor-neoantigen expression, processing, or presentation impairments as determinants of acquired ICI resistance. There is also some evidence of activation of alternative checkpoints, such as T-cell immunoglobulin and mucin domaincontaining protein 3 (TIM-3), which circumvent the blockade of PD-1/PD-L1 and CTLA-4 with ICIs in tumors with acquired resistance.
Some factors that contribute to primary resistance may also appear in acquired resistance. For example, mutations in JAK1/2 have been described in the context of both primary and acquired resistance to ICIs. This is not surprising, as JAK1/2 mutations render cells unable to respond to IFN-γ, essentially resulting in the absence of PD-L1 expression and, hence, leaving cells unresponsive to PD-1 blockade therapy.
Brahmer noted that JAK1/2 mutations appear to be common in the context of ICI resistance. Mutations in the STK11 gene also appear to be common data from a recent genomic profiling study of patients with KRAS-mutant lung adenocarcinoma identified STK11 modifications as the most prevalent genomic driver of resistance to inhibitors of the PD-1 axis.15
Future Landscape for ICIs in Immunotherapy
“We are starting to understand the origins of immunotherapy resistance and, potentially, how to overcome at least some of the mechanisms that drive this resistance,” Curran said. “The challenge [ahead] is identifying the actual mechanisms of resistance operating in a particular patient with cancer, so that a rational combination can be applied to reverse the particular [mechanisms] that dominate the resistance profile in that individual patient.”
The multiplicity and overlap between the mechanisms of resistance would require updated and rapid methods to conduct immune surveillance in individual patients based on imaging, genomic, molecular, and cellular analytics, as well as the translation of these data to algorithms for treatment combinations and agent selection to maximize the benefit to the patient.
For instance, the availability of a clinically useful, real-time systemic imaging with a whole-body hypoxia PET tracer across tumors could help determine the extent and uniformity of hypoxia in tumors and drive clinical decision making, such as monitoring of hypoxia-targeted drugs and choice and timing of immunotherapy. Such imaging agents could also help delineate the degree to which hypoxia contributes to mixed response, in which some lesions respond and others do not, Curran explained.
The rational combination of immunotherapies and/or other agents based on immune surveillance data is another area of active and exciting research in ICI therapy. Some ongoing clinical trials are addressing this based on the patient’s genetic background, Brahmer said. The HUDSON trial (NCT03334617), for example, is assessing the efficacy, safety, and tolerability of multiple biomarker-directed treatments in patients with metastatic NSCLC who have progressed on an antiPD-1/PD-L1 therapy.
There are challenges going forward, Brahmer said: “The field is rapidly evolving, and the technology needs to improve so that we can assess the immune system, the TME, and tumor itself to improve response rates and patient outcomes.
References:
- Yan Y, Kumar AB, Finnes H, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. 2018;9:1739. doi: 10.3389/fimmu.2018.01739.
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707-723. doi: 10.1016/j.cell.2017.01.017.
- O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71-81. doi: 10.1016/j.ctrv.2016.11.007.
- Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9-16. doi: 10.1038/ bjc.2017.434.
- Ho PC, Bihuniak JD, Macintyre AN, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162(6):1217-1228. doi: 10.1016/j.cell.2015.08.012.
- Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229- 1241. doi: 10.1016/j.cell.2015.08.016.
- Zhang Q, Helfand BT, Carneiro BA, et al. Efficacy against human prostate cancer by prostate-specific membrane antigen-specific, transforming growth factor-β insensitive genetically targeted CD8+ T-cells derived from patients with metastatic castrate-resistant disease. Eur Urol. 2018;73(5):648-652. doi: 10.1016/j.eururo.2017.12.008.
- Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene. 2017;36(4):439-445. doi: 10.1038/onc.2016.225.
- Jayaprakash P, Ai M, Liu A, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. 2018;128(11)5137-5149. doi: 10.1172/JCI96268.
- Weber R, Fleming V, Hu X, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. 2018;9:1310. doi: 10.3389/fimmu.2018.01310.
- Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435-5440. doi: 10.1158/0008-5472.CAN-12-0569.
- Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3.
- Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8+ T Cells to protect tumour cells. Nat Commun. 2018;9(1):948. doi: 10.1038/s41467-018-03347-0.
- Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7(3):264-276. doi: 10.1158/2159-8290.CD-16-0828.
- Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822-835. doi: 10.1158/2159-8290.CD-18-0099.
















Bispecific Antibodies and ADCs Deliver a Futuristic Horizon Across Lung Cancer Settings
October 23rd 2024Recent advancements in protein engineering, especially antibody-drug conjugates, show promise in lung cancer treatment, with ivonescimab outperforming pembrolizumab in PD-L1-positive advanced non-small cell lung cancer.
Read More


