|Videos|May 14, 2020
Transformation of Essential Thrombocythemia
Author(s)Targeted Oncology
Advertisement
Episodes in this series

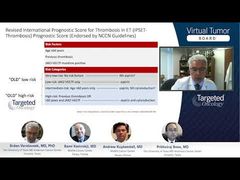
Srdan Verstovsek, MD, PhD: The progression of ET to myelofibrosis [MF] or acute myeloid leukemia [AML] is not seen very often. But it is seen, and obviously it’s a very important problem because that means a shorter life expectancy. Prithviraj, in terms of management of progression, we all agree on bone marrow biopsy, and then what happens when you have a case of elevated blasts or fibrosis with progression in the cytopenias or a big spleen? How do we go about managing post-ET [essential thrombocythemia]MF or AML? Is there any specific difference, in other words?
Prithviraj Bose, MD: I think the management of post-ET MF would be similar to that of primary MF or post-PV [polycythemia vera] MF. I think you’re still looking primarily at the spleen and symptoms as you make a decision for ruxolitinib, for example, or as you make a decision for an anemia-directed therapy. Again, you’re really looking at the picture of the whole patient, who can have not just 1 thing, but you try to choose the predominant problem as Rami alluded to earlier during his talk—to try and manage the disease. So, I think it’s either anemia-directed therapy or spleen-and-symptoms-directed therapy, which, of course, tends to be ruxolitinib. But, of course, the prognosis changes in a significant way, and I usually have that discussion with the patient when it is post-ET MF.
AML, of course, is a whole different cup of tea. When the blasts are increasing, you’re getting into an accelerated phase even before the formal post-MF, post-ET AML, you want to introduce the hypomethylating agents, for example. Those are the cornerstones right there for those patients, while perhaps continuing ruxolitinib if indicated for spleen symptoms, etc.
Srdan Verstovsek, MD, PhD: Andrew, Prithviraj was talking about when you change to myelofibrosis, then you manage basically like any other patient with myelofibrosis—looking at the symptoms, the spleen, anemia, and you may include a therapy with JAK inhibitors. Is there a role for JAK inhibitors in ET? I know there are some studies and it’s not officially approved, but is there a role or possible path forward for development? What’s your feeling about that?
Andrew Kuykendall, MD:I think that it’s less clear, obviously, than with polycythemia vera and with myelofibrosis, and probably because ET is just a little bit lower on the spectrum as far as the symptom burden. There’s less incidence of splenomegaly, and there’s less need to control the hematocrit level. So, the things that ruxolitinib have been shown to do effectively aren’t present as frequently in those patients with ET. But that doesn’t mean that none of those things is present in every patient with ET, and certainly there are some patients with ET who have significant constitutional symptoms and splenomegaly. Especially even in the patients with ET who have a JAK2 mutation, they may have hemoglobin levels that are getting close to that, that would be considered to be polycythemia vera. So, I think there is a subset of patients who could certainly benefit from ruxolitinib because we know that it’s a very good drug in controlling symptoms, reducing spleen size, and controlling hematocrit. Although the majority of patients with ET don’t need all of those things done, some do.
Srdan Verstovsek, MD, PhD: That was a really nice discussion. Thank you all very much for the review of ET. We’ll move on to the next case.
Transcript edited for clarity.
Advertisement

































Advertisement
Advertisement
Trending on Targeted Oncology - Immunotherapy, Biomarkers, and Cancer Pathways
1
Improving Survival, Quality of Life, and Cost-Effectiveness for Allo-HCT
2
FDA Panel Votes 10-3 In Favor of RP1 Plus Nivolumab for Advanced Melanoma
3
FDA Approves Lutetium Lu 177 Vipivotide Tetraxetan Plus ARPI in mAPMN/S Prostate Cancer
4
NCCN Lists Gedatolisib Regimens as Preferred Option in HR+/HER2– Metastatic Breast Cancer
5